Chapter 3. The CHM13 Cell Line
To understand why the T2T project used the CHM13 cell line, we need to understand a fundamental challenge in genome sequencing: heterozygosity.
In a typical human genome, you inherit one set of chromosomes from your mother and another from your father. These two sets are similar but not identical - they differ at millions of positions, creating what we call heterozygous variants. For most of the genome, this isn’t a problem. But in highly repetitive regions like centromeres or ribosomal DNA arrays, these small differences create a serious headache for genome assembly.
Here’s why: when you sequence DNA, you break it into millions of fragments and then use computers to reassemble them, like putting together a jigsaw puzzle. If you have two slightly different copies of a repetitive region - one from mom, one from dad - the computer can’t always tell which fragments belong to which copy. Should this read go with the maternal chromosome or the paternal one? In repetitive regions where the same sequence appears dozens or hundreds of times, this ambiguity makes accurate assembly nearly impossible.
The earlier human reference genome, GRCh38, was constructed from bacterial artificial chromosomes (BACs) - large pieces of DNA (100-200 kb) cloned into bacteria and then sequenced using Sanger technology. These BACs were opportunistically assembled from multiple individuals, creating a mosaic of haplotypes (different genetic variants from different people). This approach resulted in about 151 million base pairs of unknown sequence (gaps), mostly in repetitive regions where the heterozygous variants and structural differences between individuals made assembly too complex with available technology.
The T2T Consortium needed a different approach. They needed DNA where both chromosome copies were identical - eliminating the confusion caused by heterozygous variants. That’s where CHM13 comes in.
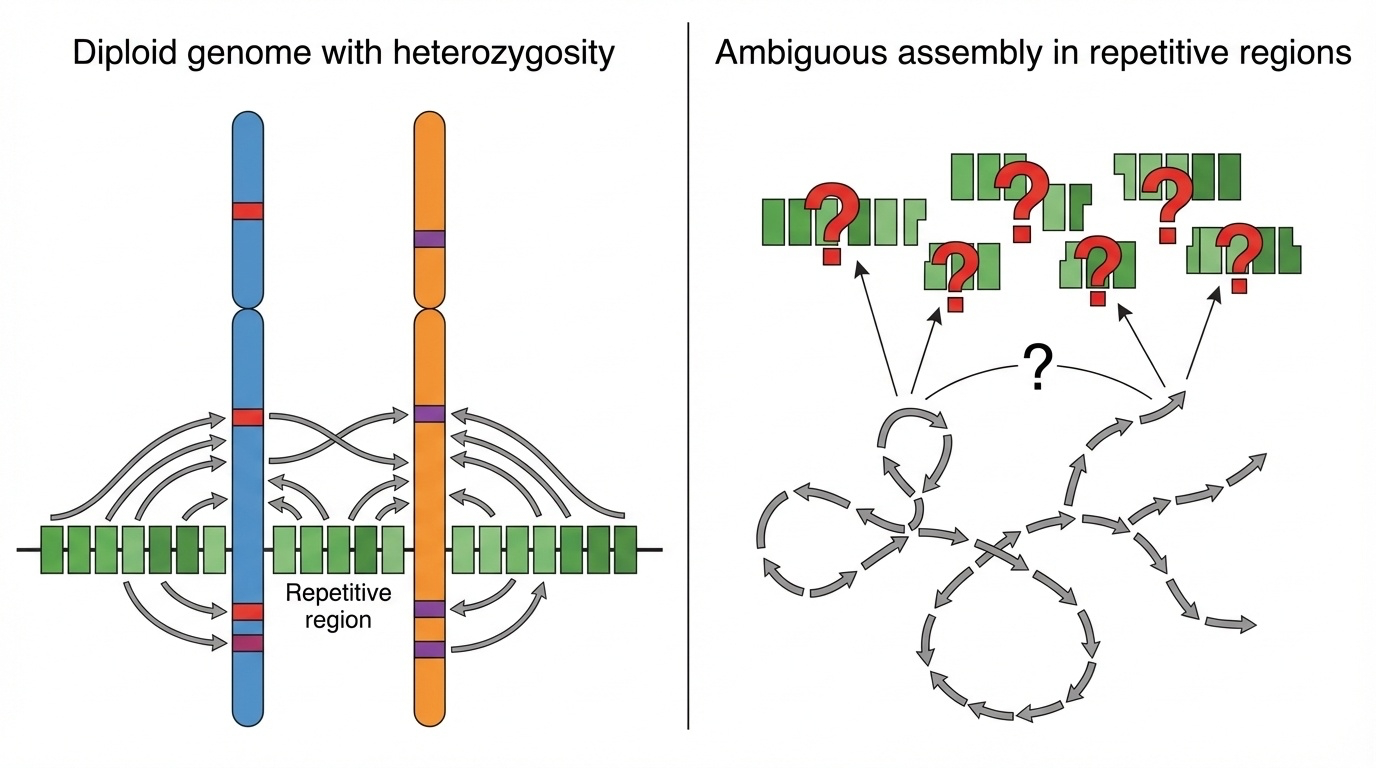
Figure 3-1: Why Heterozygosity Breaks Genome Assembly. In normal diploid genomes, heterozygosity creates ambiguity during assembly of repetitive regions. Sequencing reads from similar but not identical chromosome copies cannot be confidently assigned, leading to assembly errors or gaps. This fundamental problem prevented complete assembly of centromeres, satellite arrays, and other highly repetitive genomic regions until the use of nearly homozygous genomes like CHM13.
What Is a Complete Hydatidiform Mole?
The CHM13 cell line comes from a rare biological phenomenon called a complete hydatidiform mole (CHM). To understand what this is, let’s first review normal fertilization.
Normal Fertilization vs CHM Formation
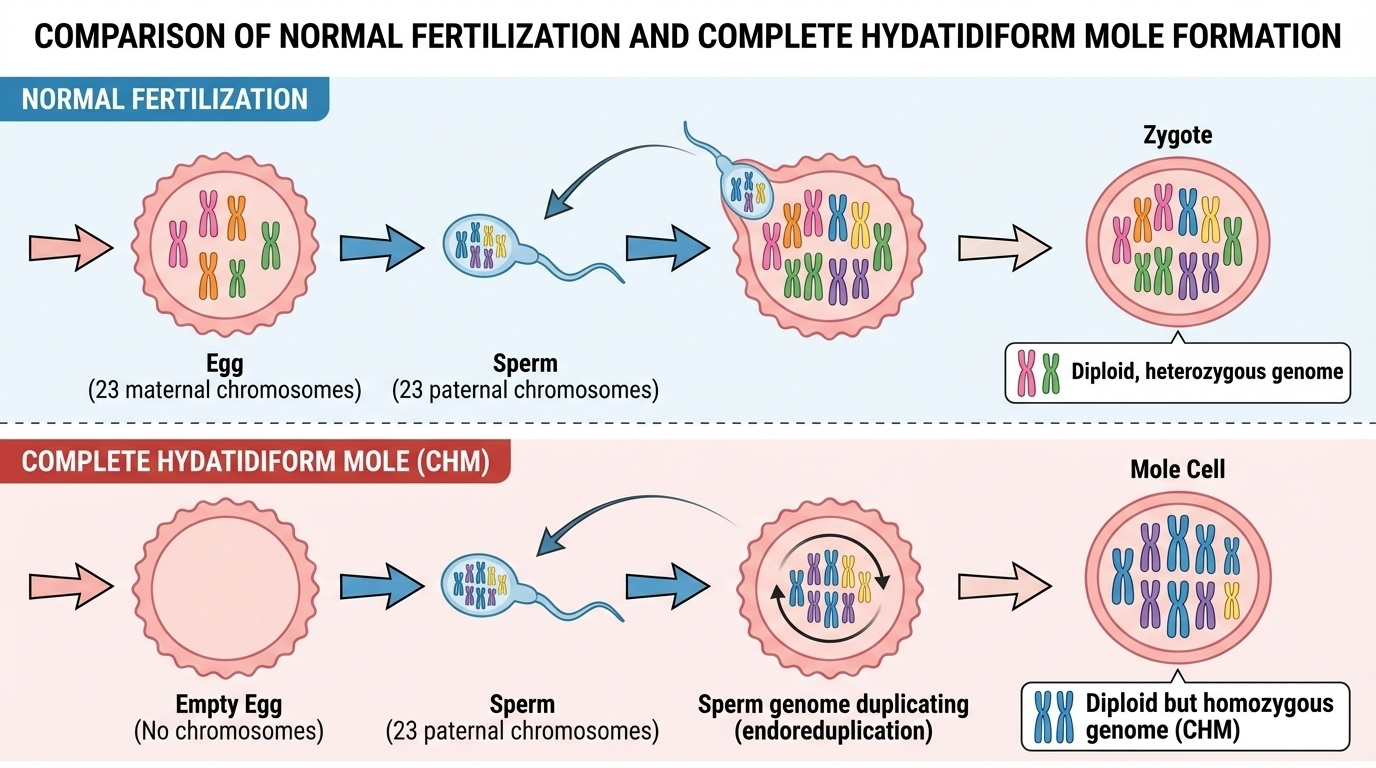
Figure 3-2: Normal Fertilization vs Complete Hydatidiform Mole. Normal fertilization combines 23 maternal chromosomes from the egg with 23 paternal chromosomes from the sperm, creating a diploid heterozygous genome. In complete hydatidiform mole formation, an empty egg (lacking maternal DNA) is fertilized by a sperm whose DNA then duplicates through endoreduplication, resulting in a diploid but homozygous genome with two identical paternal chromosome sets.
Normal Fertilization
When a sperm fertilizes an egg, the resulting cell - the zygote - has 46 chromosomes: 23 from the mother (in the egg) and 23 from the father (in the sperm). Each chromosome from mom pairs with the corresponding chromosome from dad (chromosome 1 from mom with chromosome 1 from dad, and so on). This creates a diploid genome with two versions of every chromosome.
Because mom and dad have different genetic variants, these chromosome pairs aren’t identical. You might inherit brown-eye variants from dad and blue-eye variants from mom. At the DNA sequence level, humans typically differ at about 1 in every 1,000 base pairs (0.1% of the genome), which translates to approximately 4-5 million heterozygous variants between any two chromosome copies in a diploid genome. This genetic diversity is normal and healthy - it’s why siblings look similar but not identical.
What Happens in a Complete Hydatidiform Mole
A complete hydatidiform mole occurs when something goes wrong during fertilization. The egg loses its nucleus (and therefore all maternal DNA), leaving essentially an empty egg with cytoplasm but no genetic material. When a sperm fertilizes this empty egg, there’s only paternal DNA present.
To compensate for the missing maternal contribution, the sperm’s DNA duplicates itself through a process called endoreduplication. The single set of 23 paternal chromosomes replicates to create 46 chromosomes - but they’re all identical copies from the father.
The result is a cell with 46 chromosomes - a normal number - but all of them are identical copies from the father. There are two copies of dad’s chromosome 1, two copies of dad’s chromosome 2, and so on. No maternal DNA at all.
In most cases, complete hydatidiform moles have a 46,XX karyotype because the duplicated sperm either carried an X chromosome that replicated, or two sperm (both carrying X) fertilized the empty egg and fused. Complete moles with 46,XY karyotypes are extremely rare because YY combinations are not viable - humans need at least one X chromosome to survive.
CHM13 has a 46,XX karyotype, with all chromosomes derived from a single father’s genome.
Understanding “Functionally Haploid”
This creates what geneticists call a functionally haploid genome. Let’s clarify this potentially confusing term:
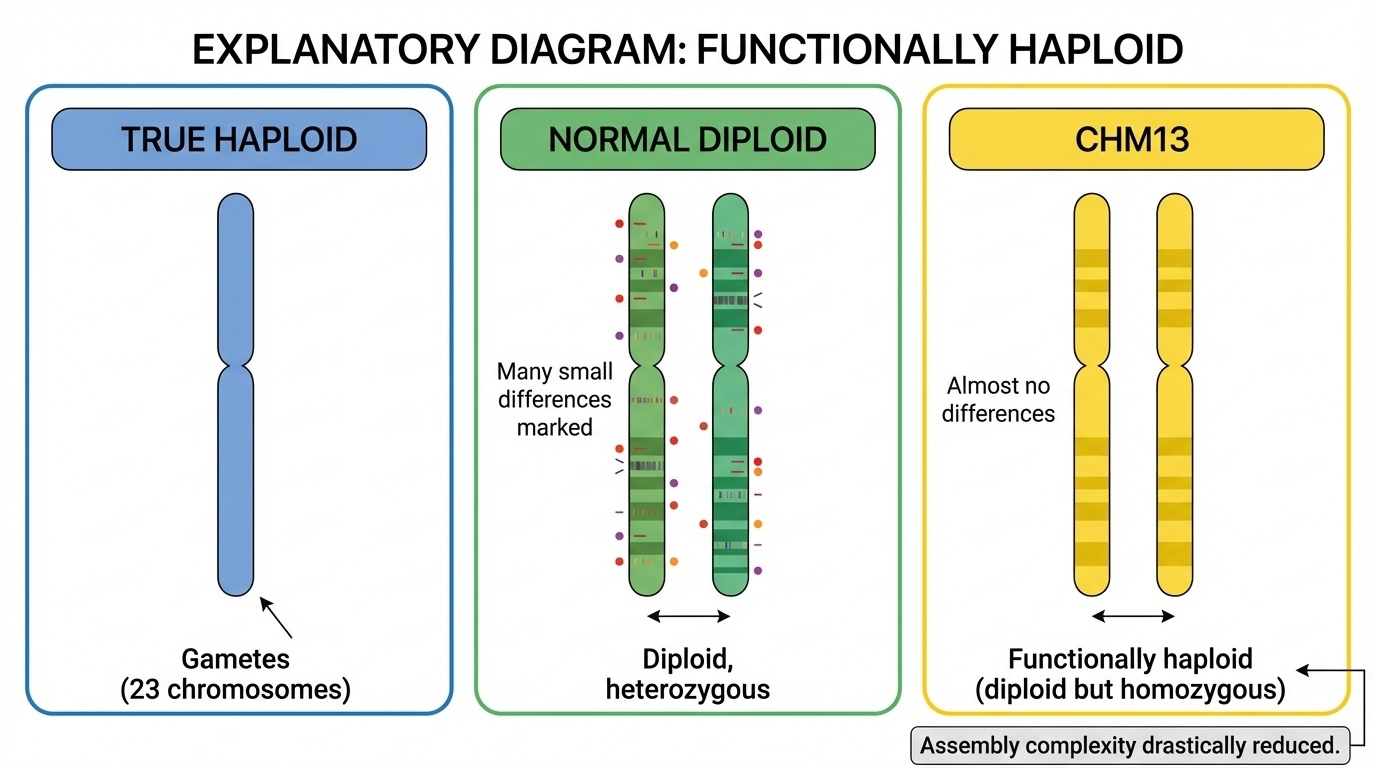
Figure 3-3: Functionally Haploid Explained. The term “functionally haploid” describes CHM13’s unique genetic state. True haploid cells (gametes) contain only 23 chromosomes. Normal diploid cells have 46 chromosomes with heterozygous differences between maternal and paternal copies. CHM13 has 46 chromosomes (diploid number) but both copies are nearly identical (homozygous), behaving like a haploid genome for assembly purposes and drastically reducing assembly complexity.
| Term | Number of Chromosomes | Are They Identical? | Heterozygous Variants | Example |
|---|---|---|---|---|
| True Haploid | 23 (one copy of each) | N/A (only one copy) | 0 (no pairs to compare) | Sperm, Egg |
| Diploid (Normal) | 46 (two copies of each) | No (maternal ≠ paternal) | ~4-5 million | You, Me, Most People |
| Functionally Haploid (CHM13) | 46 (two copies of each) | Yes (both from duplicated paternal DNA) | ~Few thousand (<0.01%) | CHM13 |
“Functionally haploid” means that even though there are two copies of each chromosome (making it diploid in chromosome number), both copies are nearly identical - so it behaves like a haploid genome for sequencing purposes. There are almost no heterozygous variants to complicate assembly.
Nearly Uniform Homozygosity
It’s important to note that CHM13 is not perfectly homozygous - no biological system is perfect. When scientists sequenced CHM13, they found:
- A few thousand heterozygous variants scattered across the genome
- One megabase-scale heterozygous deletion within the rDNA array on chromosome 15
These likely arose from:
- Rare errors during the endoreduplication process that created the mole
- Somatic mutations that occurred as the cell line was cultured over decades
However, these variants represent less than 0.01% of the genome. For comparison:
- Normal diploid genome: ~4-5 million heterozygous variants
- CHM13: ~few thousand heterozygous variants
- Reduction in heterozygosity: >99.99%
So while CHM13 isn’t perfectly homozygous, it’s close enough - the assembly is simplified by more than 99.99% compared to a typical diploid genome, which was exactly what the T2T project needed to resolve repetitive regions.
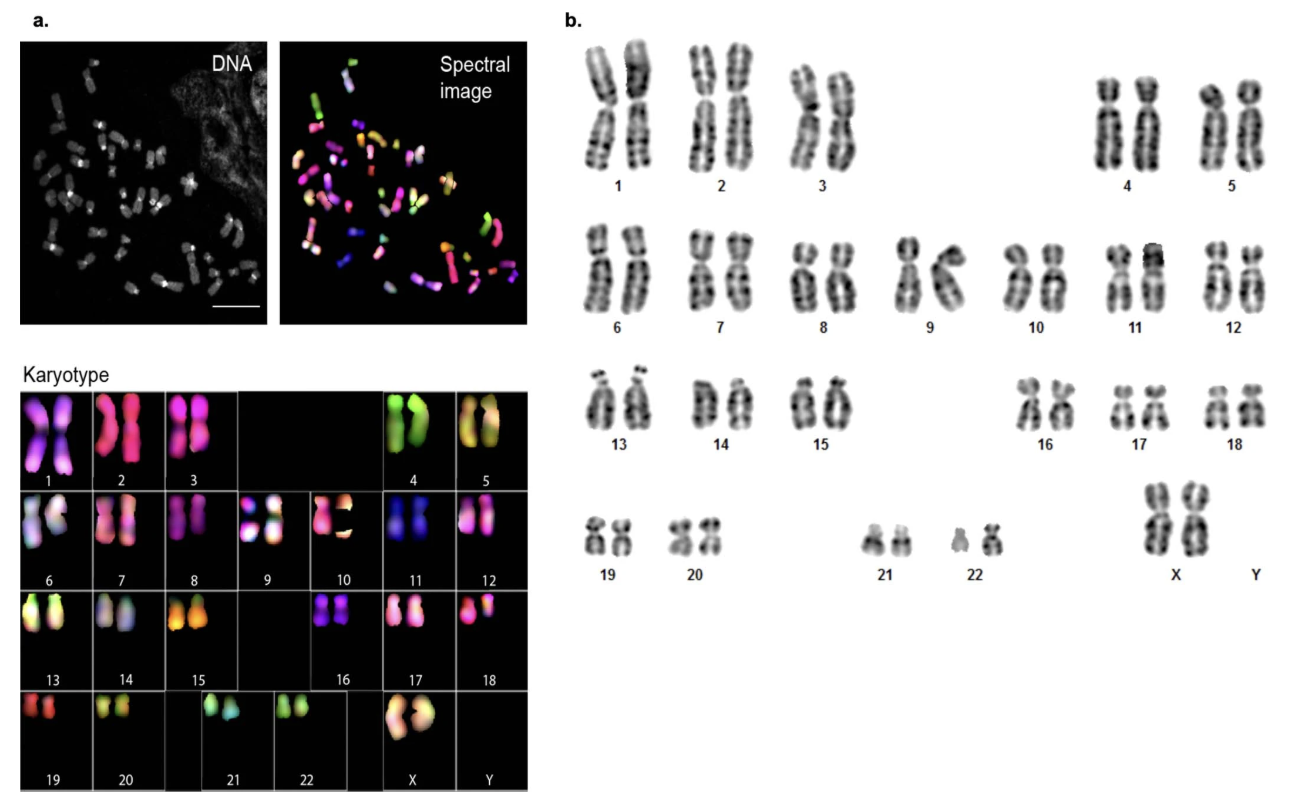
Figure 3-4: Karyotyping of CHM13. The image shows two types of chromosome analysis: (a) Spectral karyotyping (SKY), where each chromosome type is labeled with a different fluorescent color, and (b) G-banding, which reveals chromosome structure through staining patterns. Both confirm that CHM13 has a normal 46,XX karyotype with no structural abnormalities such as translocations, deletions, or duplications. Source: Miga, K.H. et al. (2020). Telomere-to-telomere assembly of a complete human X chromosome. Nature, 585, 79-84. https://doi.org/10.1038/s41586-020-2547-7. License: CC-BY 4.0.
Why This Is Perfect for Sequencing
Because both chromosome copies in CHM13 are nearly identical, there are virtually no heterozygous variants to confuse the assembly process. When sequencing reads come from a repetitive region, the computer doesn’t have to distinguish which chromosome copy they belong to - they’re essentially the same either way.
Think of it this way: imagine you’re assembling two jigsaw puzzles simultaneously.
In a normal diploid genome: You have pieces from two similar but not identical puzzles mixed together - some pieces fit only in puzzle A, some only in puzzle B, and in repetitive regions (like a solid blue sky in both puzzles), many pieces might fit in either puzzle. The assembler must figure out which pieces belong to which puzzle, which becomes nearly impossible when you have hundreds of nearly identical pieces.
In CHM13: Both puzzles are essentially identical, so it doesn’t matter which piece goes where - they fit the same pattern. This makes the assembly dramatically simpler, especially for the repetitive regions that plagued earlier sequencing efforts.
This advantage is critical for regions like centromeres, where a 2-kb repeat might be copied 1,400 times. In a diploid genome, if mom’s centromere has 1,387 copies and dad’s has 1,421 copies, with slight sequence variations between them, the assembler faces an impossible task of separating and reconstructing both. In CHM13, both copies have exactly the same number of repeats with virtually identical sequences.
The hTERT Modification: Making Cells Immortal
The full name of the cell line is CHM13hTERT. The “hTERT” part refers to a genetic modification that was added to make the cells immortal - capable of dividing indefinitely in culture.
The Telomere Problem
Normal human cells can only divide a limited number of times - typically 40 to 60 divisions (known as the Hayflick limit) - before they stop. This limit exists because of structures called telomeres, which are repetitive DNA sequences (TTAGGG repeated thousands of times) that cap the ends of chromosomes, like the plastic tips on shoelaces.
Every time a cell divides and copies its DNA, the telomeres get slightly shorter. This happens because of the end-replication problem: the DNA copying machinery (DNA polymerase) can’t fully replicate the very ends of linear chromosomes. Each cell division results in the loss of approximately 50-200 base pairs from the telomeres.

Figure 3-5: Telomere Shortening Over Cell Divisions. With each cell division, telomeres progressively shorten due to the end-replication problem. Starting with long protective caps, telomeres gradually lose 50-200 base pairs per division. After approximately 40-60 divisions (the Hayflick limit), telomeres become critically short, triggering cellular senescence and preventing further division. This progressive shortening serves as a molecular clock limiting the replicative lifespan of normal somatic cells.
Eventually, after many divisions, telomeres become so short that the cell recognizes them as damaged DNA and stops dividing - a state called senescence (cellular aging). If cells try to divide past this point, they can undergo programmed cell death (apoptosis).
This built-in limit is actually a protective mechanism that prevents damaged cells from dividing uncontrollably and potentially becoming cancerous. But for research purposes, it’s a problem. If you’re trying to grow cells for extensive sequencing experiments that require billions of cells over multiple years, you need a large, consistent supply of cells with identical DNA. Running out of cells halfway through your project isn’t an option.
Enter Telomerase
The solution is an enzyme called telomerase, which adds new TTAGGG repeats to telomeres, compensating for the bases lost during each cell division and preventing telomere shortening.
Telomerase consists of two key components:
- TERT (Telomerase Reverse Transcriptase): The catalytic protein that adds DNA repeats
- TERC (Telomerase RNA Component): An RNA template that specifies the TTAGGG sequence
Telomerase is naturally active in:
- Germ cells (sperm and eggs): Need to divide many times and pass on full-length telomeres to offspring
- Stem cells: Need to replenish tissues throughout life
- Some highly proliferative tissues: Such as cells in the bone marrow or gut lining
But telomerase is turned off in most adult somatic cells, which is why they have limited lifespans and eventually senesce.
Importantly, approximately 85-95% of cancer cells reactivate telomerase, allowing them to divide without limit - one key reason tumors can grow indefinitely. The remaining 5-15% of cancers use an alternative mechanism called ALT (Alternative Lengthening of Telomeres).
The hTERT Gene
The CHM13 cells were genetically modified to express human telomerase reverse transcriptase (hTERT), the key catalytic component of telomerase. This modification was done by introducing the TERT gene into the cells using a viral vector, causing the cells to produce telomerase continuously.
This modification makes the cells immortal - they can divide indefinitely while maintaining stable telomeres and consistent DNA. Critically, the hTERT modification only affects telomere maintenance; it doesn’t change the sequence of the 24 chromosomes (22 autosomes + X + X) that the T2T project was trying to sequence.
For the T2T project, this meant:
- Researchers could grow as many CHM13 cells as needed over the multi-year project
- They could extract large quantities of identical DNA for multiple rounds of sequencing
- The cells remain genetically stable through many passages
- The DNA sequenced at the beginning of the project matches the DNA sequenced years later
The original CHM13 cell line was developed in 1993 from a complete hydatidiform mole and has been used in genomics research for decades. The hTERT modification was added later to ensure long-term availability for research projects like T2T.
Genetic Stability and Quality Control
One major concern when using any cell line for reference genome sequencing is genetic stability. Cells grown in culture for many generations can accumulate mutations, chromosomal rearrangements, or other changes that would compromise the reference genome.
The T2T Consortium performed extensive quality control to ensure CHM13’s stability:
Karyotype Analysis
G-banding and spectral karyotyping (SKY) were performed to visualize all 46 chromosomes and check for:
- Correct chromosome number (should be 46)
- Proper chromosome structure (no translocations, deletions, or duplications)
- No evidence of aneuploidy (extra or missing chromosomes)
Results confirmed a normal, stable 46,XX karyotype with no observable chromosomal abnormalities (Extended Data Fig. 1 from Miga et al. 2020).
Sequence-Level Validation
The assembly itself provided evidence of genetic stability:
- Uniform read coverage across all chromosomes (no large deletions or duplications)
- Low heterozygosity confirmed throughout the genome (~few thousand variants, as expected)
- Consistent sequence quality across multiple sequencing runs over several years
Why These Checks Matter
Unlike cancer cell lines (which also divide indefinitely through telomerase or ALT), CHM13hTERT maintains:
- Normal chromosome structure: No translocations or aneuploidy
- Genetic stability: Minimal mutations accumulating over time
- Reproducible results: The same DNA sequence across multiple years and experiments
This stability is crucial for creating a reference genome. If the cells were accumulating mutations or chromosomal changes, the reference would be unreliable.
Ancestry and Genetic Background
The CHM13 genome doesn’t represent all of humanity - it comes from a single source of DNA (one father’s duplicated genome). Genetic ancestry analysis was performed using maximum likelihood admixture analysis to understand the genetic background of CHM13.

Figure 3-6: Genetic Ancestry Analysis of CHM13. Admixture analysis reveals the genetic ancestry composition of the CHM13 cell line. The analysis shows CHM13 has predominantly European ancestry, with smaller contributions from other ancestral populations. This ancestry information is important for understanding potential biases in using CHM13 as a reference genome and for comparative genomic studies across diverse human populations. Source: Miga, K.H. et al. (2020). Telomere-to-telomere assembly of a complete human X chromosome. Nature, 585, 79-84. https://doi.org/10.1038/s41586-020-2547-7. License: CC-BY 4.0.
The analysis revealed:
- Primarily European ancestry (approximately 70-80% of the genome)
- Small amounts of admixture: Possible South Asian, East Asian, or Native American ancestry
- Traces of Neanderthal DNA: Like most non-African modern humans, CHM13 carries approximately 1-2% Neanderthal DNA, inherited from interbreeding that occurred roughly 50,000-60,000 years ago
Does Ancestry Matter?
For the T2T project’s goals, the specific ancestry of CHM13 doesn’t significantly limit its usefulness. Here’s why:
What matters about CHM13:
- Lack of heterozygosity → enables complete assembly of repetitive regions
- Complete structural template → shows where everything is located
- Gap-free reference → resolves previously unsequenced regions
What doesn’t matter as much:
- Specific genetic variants → these vary between individuals anyway
- Population-specific alleles → the pangenome captures these
- Ancestry composition → any single genome has limitations
Think of it like creating the first complete map of a city’s streets. It doesn’t matter who drew the map or where the cartographer came from - what matters is that now we can see all the streets, buildings, and landmarks that were previously unmapped. Once we have that complete map, we can study how traffic patterns (genetic variants) differ in different neighborhoods (populations).
The key advantage of CHM13 isn’t its specific genetic variants, but rather its homozygous structure, which enabled complete assembly of complex repetitive regions. Once you have that complete structural map, you can study how different variants exist in different populations - which is exactly what the Human Pangenome Reference Consortium (HPRC) is doing.
Important context: The limitation isn’t CHM13’s European ancestry specifically, but rather that any single genome can’t represent all of humanity. Even if we had chosen a complete genome from an African, Asian, or South American individual, it still wouldn’t capture human diversity. That’s why we need pangenome references.
Why CHM13 Specifically?
You might wonder: if complete hydatidiform moles create homozygous genomes, why not use any CHM cell line? Why CHM13 specifically?
Several factors made CHM13 the ideal choice:
1. Availability and Characterization
CHM13 was already well-characterized and available to researchers. It had been used in previous genomics studies since the 1990s, including:
- Segmental duplication analysis
- As a benchmark genome for variant calling (part of the Genome in a Bottle consortium)
- Validating assembly methods
2. Karyotype Stability
Unlike some cell lines that accumulate chromosomal abnormalities in culture, CHM13 maintained a stable, normal 46,XX karyotype through many passages over decades. This stability is unusual and valuable.
3. Favorable Karyotype (46,XX)
Having two X chromosomes (rather than XY) meant the project could immediately work on completing the X chromosome, which:
- Had been extensively studied
- Was chosen as the first target (completed in 2020)
- Provided proof of concept for the methods
4. Successful hTERT Immortalization
The hTERT modification worked well in this line, ensuring:
- Unlimited cell availability
- Genetic stability despite unlimited passages
- No chromosomal abnormalities from the immortalization
5. High-Quality DNA
CHM13 cells produce high-quality, high-molecular-weight DNA suitable for:
- Ultra-long-read Nanopore sequencing (reads >100 kb, with some >1 Mb)
- PacBio HiFi sequencing
- Multiple complementary technologies
6. Community Support
By 2017, when the T2T project began seriously, CHM13 was already:
- Used by multiple research groups
- Part of the Genome in a Bottle reference materials
- Well-documented with shared protocols
Other CHM cell lines exist, but CHM13 had the right combination of properties, existing characterization, stability, and research community support to serve as the foundation for the T2T project.
Why CHM13 Made T2T Possible
Let’s bring all these pieces together. The T2T Consortium needed to sequence regions that had defeated earlier efforts centromeres (multi-megabase satellite arrays), ribosomal DNA arrays (hundreds of tandem repeats), segmental duplications (>90% sequence identity), and heterochromatic regions like Yq12 (>30 Mb of satellites). These regions are highly repetitive, and in a normal diploid genome, small differences between the two chromosome copies make accurate assembly nearly impossible.
The CHM13hTERT cell line solved this problem through three key features:
1. Functionally Haploid Genome
Both chromosome copies are nearly identical (duplicated paternal DNA), eliminating >99.99% of heterozygous variants that confuse assembly algorithms in repetitive regions. This single feature enabled assembly of:
- All 24 centromeres completely (ranging from 366 kb to several Mb)
- Short arms of acrocentric chromosomes (66.1 Mb total)
- Complex segmental duplications (6.61% of genome vs 5.00% in GRCh38)
- Y chromosome heterochromatin (>30 Mb, added from HG002 in v2.0)
The few thousand remaining heterozygous variants (~0.01% of genome) were manageable compared to the 4-5 million variants in a normal diploid genome.
2. Immortalized Cells
The hTERT modification allows unlimited cell division, providing a consistent, stable supply of DNA for extensive sequencing. The T2T project required enormous amounts of high-quality DNA:
- 30× PacBio HiFi sequencing (~20 kb reads, high accuracy)
- 50× Oxford Nanopore ultra-long-read sequencing (>100 kb reads, up to 1+ Mb)
- 100× Illumina short-read sequencing (for polishing)
- Additional data: Hi-C (chromatin conformation), Bionano optical mapping (physical map), Strand-seq (detecting inversions)
Generating this much data across multiple technologies over several years required a cell line that could provide unlimited, consistent DNA - which CHM13hTERT delivered.
3. Genetic Stability
Karyotype analysis confirmed a normal 46,XX chromosome structure with no abnormalities, ensuring the cells remain stable through many passages. Unlike some cancer cell lines (which also divide indefinitely), CHM13hTERT maintains:
- Normal chromosome number
- No translocations or large structural variants
- Minimal accumulation of mutations over time
This stability was critical for generating consistent data across the entire multi-year project timeline.
When combined with advanced long-read sequencing technologies and innovative computational methods (marker-assisted polishing, string graph assembly), CHM13’s unique properties enabled the first complete human genome assembly - all 3.055 billion base pairs with no gaps, covering the 22 autosomes and the X chromosome. The Y chromosome was added later from a different genome (HG002, a male genome commonly used for benchmarking) to create T2T-CHM13v2.0.
This achievement wouldn’t have been possible with DNA from a typical diploid individual. Even with today’s best long-read technologies, assembling a diploid human genome to telomere-to-telomere completeness remains challenging because of the need to separate and assemble two similar but distinct haplotypes in repetitive regions.
CHM13 provided the genomic simplicity needed to finally see what was hiding in the genome’s most complex regions.
Comparing Haploid and Diploid Approaches
The table below summarizes how CHM13’s functionally haploid genome compares to the approaches used in earlier reference genomes like GRCh38.
| Feature | CHM13 (Functionally Haploid) | GRCh38 (Diploid, Mosaic) |
|---|---|---|
| Source | One complete hydatidiform mole (duplicated paternal DNA) | Multiple individuals, BAC clones from different donors |
| Chromosome Copies | Two nearly identical copies from duplicated paternal DNA | Mosaic of sequences from multiple individuals; represents different haplotypes |
| Heterozygous Variants | ~Few thousand (<0.01% of genome) | ~4-5 million per individual; structural differences between BAC clone donors |
| Assembly Complexity | Simple - minimal ambiguity in repetitive regions | Complex - heterozygous variants and structural differences create ambiguity, especially in repeats |
| Gaps in Assembly | Zero gaps - complete 3.055 billion bp coverage (with Y from HG002 in v2.0) | ~151 million bp of unknown sequence (N-bases), mostly in repetitive regions |
| Centromeres | Fully assembled - all 24 centromeres complete, base-to-base | Largely absent or represented by small consensus sequences (~few kb placeholders) |
| Segmental Duplications | 6.61% of genome (201.93 Mb) accurately assembled | 5.00% - many regions collapsed, incorrectly assembled, or missing |
| Acrocentric Short Arms | Complete - 66.1 Mb including all rDNA arrays | Almost entirely absent - represented as gaps |
| Y Chromosome | Complete in v2.0 from HG002 (62.46 Mb, no gaps) | >50% missing in GRCh38-Y (represented as one 30+ Mb gap) |
| Accuracy | 99.99% (1 error per 10 million bases) in unique regions; ~99.3% in largest satellite arrays | Very high in non-repetitive regions; undefined/not measurable in gaps |
| Technology | Long-read sequencing (PacBio HiFi, ONT ultra-long) + polishing | BAC cloning + Sanger sequencing (technology of early 2000s) |
| Assembly Approach | String graphs, marker-assisted polishing, ultra-long reads | Clone-by-clone BAC sequencing, short-read overlap assembly |
| Strengths | - Complete structural template - Enables studies of centromeres, satellites, and complex repeats - No reference gaps - High accuracy even in difficult regions |
- Represents some genetic diversity (though limited) - Historical milestone - Mosaic includes some variant information - Well-validated in non-repetitive regions |
| Limitations | - Single genome source (one duplicated paternal genome) - Doesn’t capture population diversity - No heterozygosity information - Chimeric (X from CHM13, Y from HG002) |
- Incomplete in repetitive regions - Mosaic of different individuals - Reference bias toward source populations - Major gaps in functionally important regions |
| Best Use Cases | - Structural reference for complete genomic architecture - Centromere and satellite DNA studies - Complete gene annotations - Understanding complex repeat organization |
- Standard variant calling in non-repetitive regions - Clinical diagnostics (with known limitations) - Many established bioinformatics workflows |
The Limitations of CHM13
While CHM13 was essential for creating the first complete human genome, it’s important to understand its limitations:
1. Represents Only One Genome Source
CHM13 is derived from a single father’s duplicated DNA. It doesn’t represent:
- Genetic diversity across human populations: Different populations have different variants, structural variants, and gene copy numbers
- The typical diploid state: Most humans have two different copies (maternal and paternal) of each chromosome
- Maternal contributions: All sequences come from one father
- Common structural variants: Many people carry structural variants (inversions, duplications, deletions) that aren’t in CHM13
2. Ancestry Bias
With primarily European ancestry, CHM13 may not optimally represent:
- Variants common in African populations: Africa has the most genetic diversity
- Asian-specific structural variants: Important variants in East Asian, South Asian, or Native American populations
- Adaptive variants: Genetic changes that helped populations adapt to different environments
- Population-specific gene families: Some genes show copy number variation between populations
3. No Heterozygosity Information
Because both chromosome copies are nearly identical, CHM13 can’t inform:
- Phasing of variants: Which variants appear together on the same chromosome (linkage)
- Compound heterozygous effects: Different mutations on each chromosome copy
- Heterozygous structural variants: Many structural variants are heterozygous in populations
4. Chimeric Reference (X and Y from Different Sources)
Because CHM13 is 46,XX:
- The Y chromosome in T2T-CHM13v2.0 comes from HG002 (a different male genome)
- This creates a chimeric reference for researchers working with both X and Y
- The Y chromosome represents a different genetic background than the rest of the genome
5. Cannot Study Allelic Imbalance
In normal diploid genomes, the two chromosome copies can have different expression levels or functions. CHM13 can’t inform:
- Allele-specific expression: Whether maternal or paternal alleles are expressed differently
- Parent-of-origin effects: Whether inheriting a variant from mom vs dad matters
- X-inactivation patterns: Normal females have one X inactivated; CHM13’s pattern may differ
Moving Beyond CHM13
While CHM13 was essential for creating the first complete human genome, it represents only one genome source - a single father’s duplicated DNA. Human genetic diversity is vast, with populations differing in thousands of structural variants, gene copy numbers, and sequence variations.
The key lesson: The limitation isn’t CHM13’s specific ancestry, but rather that any single genome can’t represent all of humanity. Even a complete genome from an African, Asian, or South American individual wouldn’t capture human diversity.
This is why the field is now moving toward pangenome references - collections of many complete, high-quality genomes that represent human diversity. The Human Pangenome Reference Consortium (HPRC) is building on the T2T foundation by sequencing hundreds of diverse individuals, creating graph-based references that show all the different “paths” through variable regions of the genome. We’ll explore this in detail in the next chapter.
For now, understand that CHM13 provided something essential: the first complete structural template of a human genome. It showed us what was hiding in the 151 million base pairs of gaps. Now we can study how these complex regions vary across populations but first, we needed to see the complete structure.
Think of it like cartography: Before mapping how every city differs, you first need to know what a complete city map looks like - including the streets that were previously unmapped. CHM13 gave us that complete map.
Summary
The CHM13hTERT cell line made the T2T project possible through three unique features:
1. Functionally haploid genome: Nearly identical chromosome copies (>99.99% homozygous) eliminated the heterozygosity problem that prevented assembly of repetitive regions.
2. Immortalized cells: The hTERT modification provided unlimited, genetically stable DNA for the multi-year, multi-technology sequencing effort.
3. Genetic stability: Normal 46,XX karyotype with no chromosomal abnormalities, confirmed through extensive validation.
These properties enabled the first complete human genome - all 3.055 billion base pairs with zero gaps - revealing ~200 million base pairs of previously unknown sequence in centromeres, rDNA arrays, segmental duplications, and heterochromatic regions.
CHM13’s value and limitations:
- Value: Provides the first complete structural template of human chromosomes
- Limitation: Represents only one genome; doesn’t capture human diversity
- Solution: Pangenome projects build on this foundation to represent diversity
CHM13 wasn’t the endpoint of genome sequencing - it was the essential foundation that makes studying human genetic diversity at complete resolution now possible.
Key Takeaways
-
Heterozygosity prevents assembly of repetitive regions - CHM13’s near-homozygosity solved this fundamental problem.
-
“Functionally haploid” means diploid in number (46 chromosomes) but homozygous in sequence (both copies nearly identical).
-
Complete hydatidiform moles form when an empty egg is fertilized by a sperm whose DNA then duplicates through endoreduplication.
-
The hTERT modification enabled unlimited cell growth without compromising genetic stability.
-
CHM13 represents one genome, not human diversity - but provided the complete structural map needed to study diversity.
-
The first complete human genome filled ~151 million base pairs of gaps and revealed the hidden biology of our most complex genomic regions.