Chapter 19. Genetic Architecture of Traits and Disorders
Interactive Experience: Genetic Architecture — Explore composite architecture, risk accumulation, and how genetic heterogeneity converges on shared pathways.
We’ve learned that GWAS can identify hundreds or even thousands of genetic variants associated with a trait. But here’s a question that might be puzzling you: if we’ve found all these variants, why can’t we fully predict who will get a disease? Why do some people with “high-risk” genes stay healthy, while others with seemingly lower genetic risk develop the condition?
The answer lies in something called genetic architecture—the full structure of genetic influences on a trait or disorder. It’s not just about counting up risk alleles. It’s about understanding how different types of variants—rare and common, large-effect and small-effect—work together. It’s about recognizing that genes act in specific tissues, at specific times, and in specific contexts. And it’s about appreciating that the same disease can arise through many different genetic routes.
In this chapter, we’ll explore the genetic architecture of three complex disorders: dilated cardiomyopathy (DCM), Alzheimer’s disease, and autism spectrum disorder (ASD). Each one tells us something different about how genetics shapes disease. By the end, you’ll understand why different people can develop the same disorder through completely different genetic mechanisms—and why those diverse mechanisms often converge on the same biological pathways.
What is Genetic Architecture?
Think of genetic architecture like the blueprint of a city. A city isn’t just buildings—it’s roads, utilities, parks, neighborhoods, all interconnected in complex ways. Some structures are huge and obvious (like a downtown skyscraper). Others are small but numerous (like individual houses). And the way the city functions depends not just on the parts, but on how they interact.
Genetic architecture is the same idea, but for traits and diseases. It encompasses all the genetic contributors to a phenotype, including:
Variant types: Not all genetic changes are the same. You might have single nucleotide variants (SNVs—single-letter changes), copy-number variants (CNVs—duplications or deletions of DNA segments), indels (small insertions or deletions), or larger structural variants. Each type can affect genes in different ways.
Effect sizes: Some variants have large, dominant effects—like the FGFR3 mutation causing achondroplasia that we discussed earlier. Others have tiny additive effects that only matter when combined with hundreds of other variants. The genetic architecture includes both ends of this spectrum.
Allele frequency: Are we talking about rare variants that appear in less than 0.1% of people, or common variants that show up in 10% or more? Rare variants tend to have bigger effects (otherwise natural selection would weed them out). Common variants usually have smaller effects but are easier to study because more people carry them.
Interactions: Genes don’t act in isolation. Gene-gene interactions (epistasis) happen when the effect of one variant depends on which alleles you have at another gene. Gene-environment interactions (G×E) occur when genetic risk is modified by environmental factors like diet, stress, or toxins.
Context dependencies: When does the gene act? During fetal development? In childhood? Only in old age? Which tissues matter? Does it work differently in males versus females? All of this context shapes how genetic variants manifest.
Modern genomics—whole-genome sequencing (WGS), large GWAS cohorts with hundreds of thousands of people—reveals that most traits and diseases follow a composite architecture. That means they’re shaped by a mixture of rare and common variants, with modifiers and interactions layered on top. There’s no single “disease gene” that explains everything. Instead, you have a complex landscape of genetic influences.
Let’s look at three examples to see how this plays out.
Example 1: Dilated Cardiomyopathy (DCM)
Dilated cardiomyopathy (DCM) is a heart condition where the left ventricle—the main pumping chamber—becomes enlarged and weakened. The heart stretches out, losing its ability to contract efficiently. Over time, this leads to heart failure.
For decades, doctors knew DCM could run in families, suggesting a genetic component. But the genetic architecture turned out to be more complex than anyone expected.
Rare High-Impact Variants
About 10–25% of DCM patients carry pathogenic mutations in key heart genes like TTN, LMNA, DSP, or FLNC (Zheng et al. 2024, Nat Genet; Hershberger et al. 2021, Circulation). These are rare variants—each mutation might appear in only one family—but they have big effects. If you carry a pathogenic TTN mutation (TTN encodes titin, a giant protein that forms the scaffold of heart muscle), your risk of developing DCM is high.
But here’s the thing: even among people with these pathogenic mutations, not everyone develops DCM. Some carriers stay healthy their whole lives. That tells us the rare mutation isn’t the whole story.
Polygenic Risk
A massive GWAS compared over 14,000 DCM cases with more than a million controls and found 80 genomic loci associated with DCM (Zheng et al. 2024, Nat Genet). These are common variants with small effects—the kind we’ve been talking about throughout this chapter.
Individually, each of these 80 loci barely nudges your risk. But collectively, they add up. When a polygenic score (PGS) for DCM was calculated, people in the top 10% of PGS had about 2.8 times the risk of those in the bottom 10%—even without any rare pathogenic mutations.
The Plot Thickens: Modifiers and Multi-Hit Models
Here’s where it gets really interesting. Among people who carry rare pathogenic variants, those with high PGS were much more likely to develop DCM than carriers with low PGS.
In other words, your polygenic background modifies the penetrance of rare mutations. The rare mutation doesn’t act alone—it’s influenced by the cumulative effect of common variants across the genome.
Some DCM patients also carry multiple rare variants in different cardiomyopathy-relevant genes (Hershberger et al. 2021, Circulation). It’s not one big hit; it’s several smaller hits that accumulate. Each variant on its own might not be enough to cause disease, but together they cross a threshold.
Limited Yield of Clinical Panels
When the evidence for about 51 candidate genes often tested in clinical genetic panels was curated, only about 19 genes had strong or moderate evidence for causing monogenic DCM (Hershberger et al. 2021, Circulation). This means that even our best-known “DCM genes” explain only a fraction of cases.
The rest? A mix of polygenic risk, rare variants in genes we don’t yet fully understand, and environmental factors like viral infections or alcohol use.
Functional Convergence
Despite all this genetic diversity, there’s a unifying theme. The genes identified in both the rare-variant studies and the GWAS converge on specific biological pathways: muscle structure (sarcomeres and cytoskeleton), cell adhesion (how heart cells stick together), and extracellular matrix (ECM) (the scaffolding that holds the heart together).
Many of these genes are highly expressed in cardiomyocytes (heart muscle cells) and fibroblasts (cells that produce the ECM). So even though patients might have different genetic variants, those variants often disrupt the same core mechanisms.
It’s like many roads leading to the same destination. Different starting points, but they all converge on heart muscle dysfunction.
Example 2: Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common cause of dementia, characterized by progressive memory loss, cognitive decline, and brain pathology including amyloid plaques and neurofibrillary tangles. Its genetic architecture is similarly complex.
Rare Mutations in Early-Onset Familial AD
A small fraction of Alzheimer’s cases—less than 1%—are caused by rare, highly penetrant mutations in three genes: APP, PSEN1, and PSEN2. These mutations cause early-onset familial AD, where symptoms appear before age 65, sometimes as early as the 30s or 40s. If you carry one of these mutations, your risk is nearly 100%. But these cases are exceptional.
The APOE ε4 Allele
The biggest genetic risk factor for late-onset AD—the common form that affects people in their 60s, 70s, and beyond—is the APOE ε4 allele. APOE comes in three major versions: ε2, ε3, and ε4. The ε4 allele increases AD risk by about 3-fold if you have one copy and 12-fold if you have two copies.
APOE ε4 is common—about 15% of people of European ancestry carry at least one copy. Unlike the rare APP or PSEN mutations, ε4 is not a guarantee of disease. Plenty of ε4 carriers never develop Alzheimer’s, and plenty of non-carriers do. It’s a strong risk factor, not a deterministic cause.
Polygenic Background
Large GWAS meta-analyses have identified many additional loci with smaller effects. These variants implicate pathways involved in immune function (especially microglia, the brain’s immune cells), lipid metabolism (how the brain handles cholesterol and other fats), and synaptic function (how neurons communicate) (Bellenguez et al. 2022, Nat Genet; Logue et al. 2023, Nat Genet).
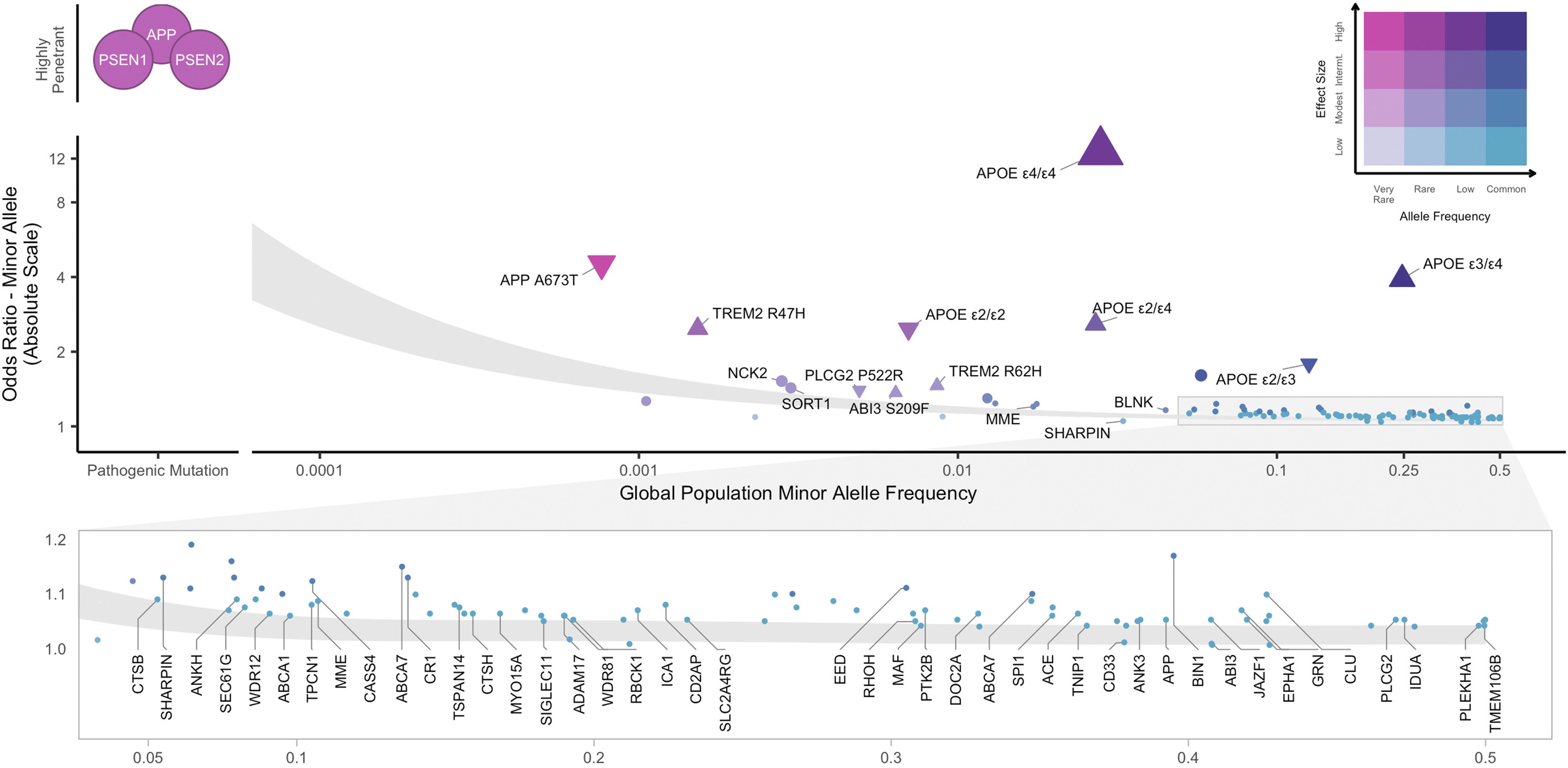
Figure: Alzheimer’s genetic landscape across the genome. This Manhattan plot shows GWAS results for Alzheimer’s disease and dementia. Each dot represents a SNP, and tall peaks indicate strong associations. Notice the peaks are scattered across many chromosomes—this is genetic heterogeneity in action. No single gene dominates. Instead, risk is distributed across the genome, with multiple loci contributing small effects that collectively shape disease susceptibility. Source: Andrews, S.J. et al. (2023). Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. EBioMedicine. https://www.thelancet.com/journals/ebiom/article/PIIS2352-3964(23)00077-0/fulltext. License: CC-BY.
Converging Pathways: Microglial Efferocytosis
Despite the genetic complexity, many of the implicated genes converge on a specific biological process: microglial efferocytosis. This is the process by which microglia—the brain’s resident immune cells—recognize and clear away dead cells and debris.
Think of microglia as the brain’s garbage collectors. They patrol the brain, looking for damaged neurons, protein aggregates (like amyloid), and other cellular trash. When they find it, they engulf it, digest it, and recycle the components. This process is called efferocytosis (literally “eating of dead cells”).
Here’s the problem: many AD risk genes—both rare and common—affect this process. TREM2, ABCA1, APOE, LPL, and others are all involved in different stages of efferocytosis:
- Detecting “eat-me” signals on damaged cells (like phosphatidylserine)
- Engulfing debris into phagosomes
- Digesting material through lysosomal fusion
- Processing metabolic byproducts like cholesterol
If this clearance system is impaired, amyloid and other toxic proteins accumulate, driving neurodegeneration.
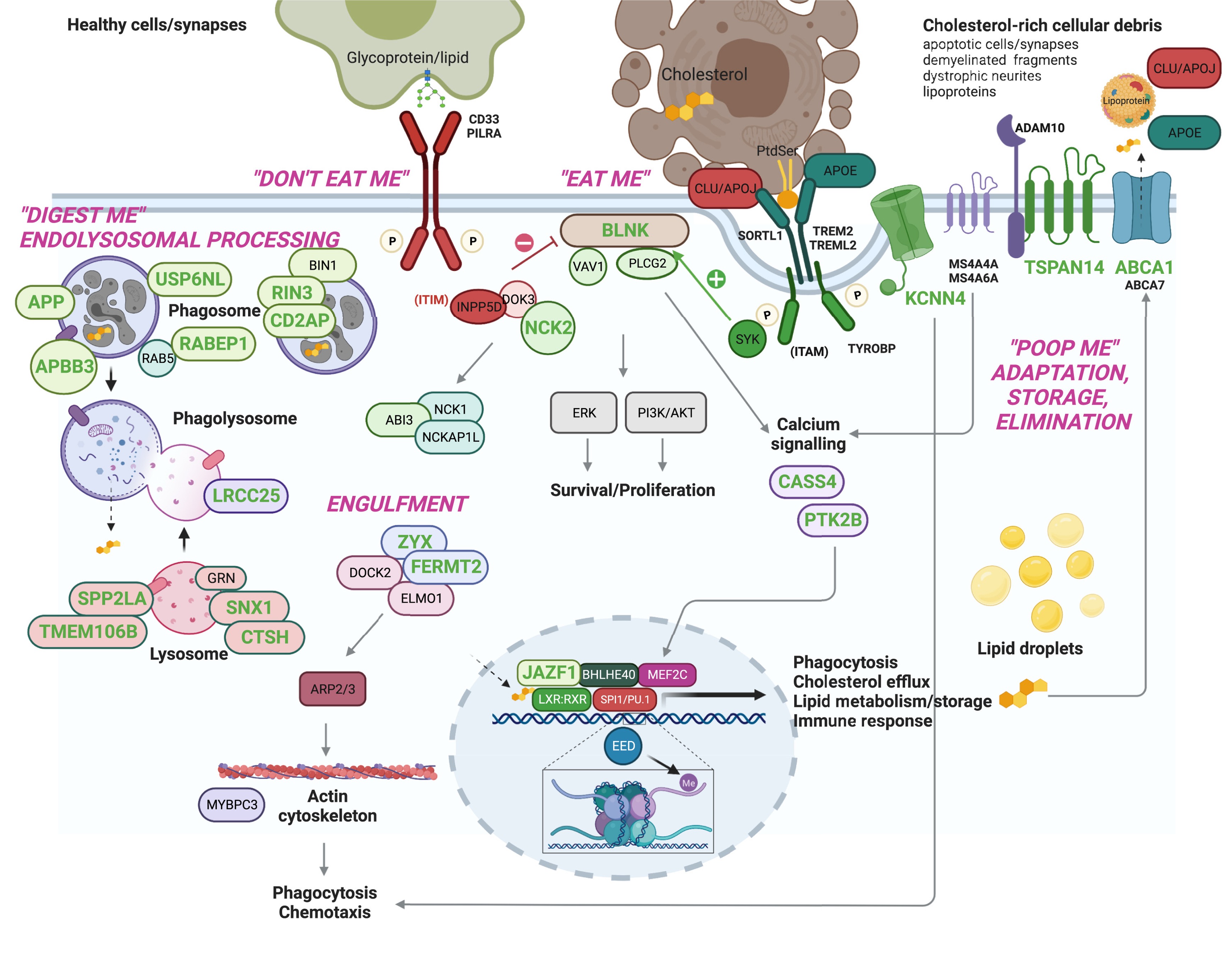
Figure: Prioritized AD risk genes converge on microglial efferocytosis. Many Alzheimer’s risk genes—both rare and common—converge on microglial efferocytosis, the process by which microglia clear dead cells and debris. Different genes affect different stages: recognizing targets (TREM2), engulfing debris, digesting it (lysosomes), and recycling components (cholesterol metabolism via ABCA1, APOE, LPL). This functional convergence suggests that impaired microglial clearance is a key hub in Alzheimer’s pathology, linking immune dysfunction, lipid metabolism, and neurodegeneration. Source: Andrews, S.J. et al. (2023). Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. EBioMedicine. https://www.thelancet.com/journals/ebiom/article/PIIS2352-3964(23)00077-0/fulltext. License: CC-BY.
Composite Model of Risk
Late-onset Alzheimer’s is influenced by a composite model: rare variants (like APOE ε4 homozygosity acts almost like a rare variant in terms of effect size), common polygenic risk, and environmental/vascular factors (hypertension, diabetes, cardiovascular health).
A recent study from the K-ROAD consortium showed that individuals with multiple genetic risk factors—for example, APOE ε4 plus high polygenic score plus rare noncoding variants or structural variants—have the highest amyloid burden and the worst cognitive outcomes. Risk accumulates (K-ROAD consortium 2025, Nat Commun).
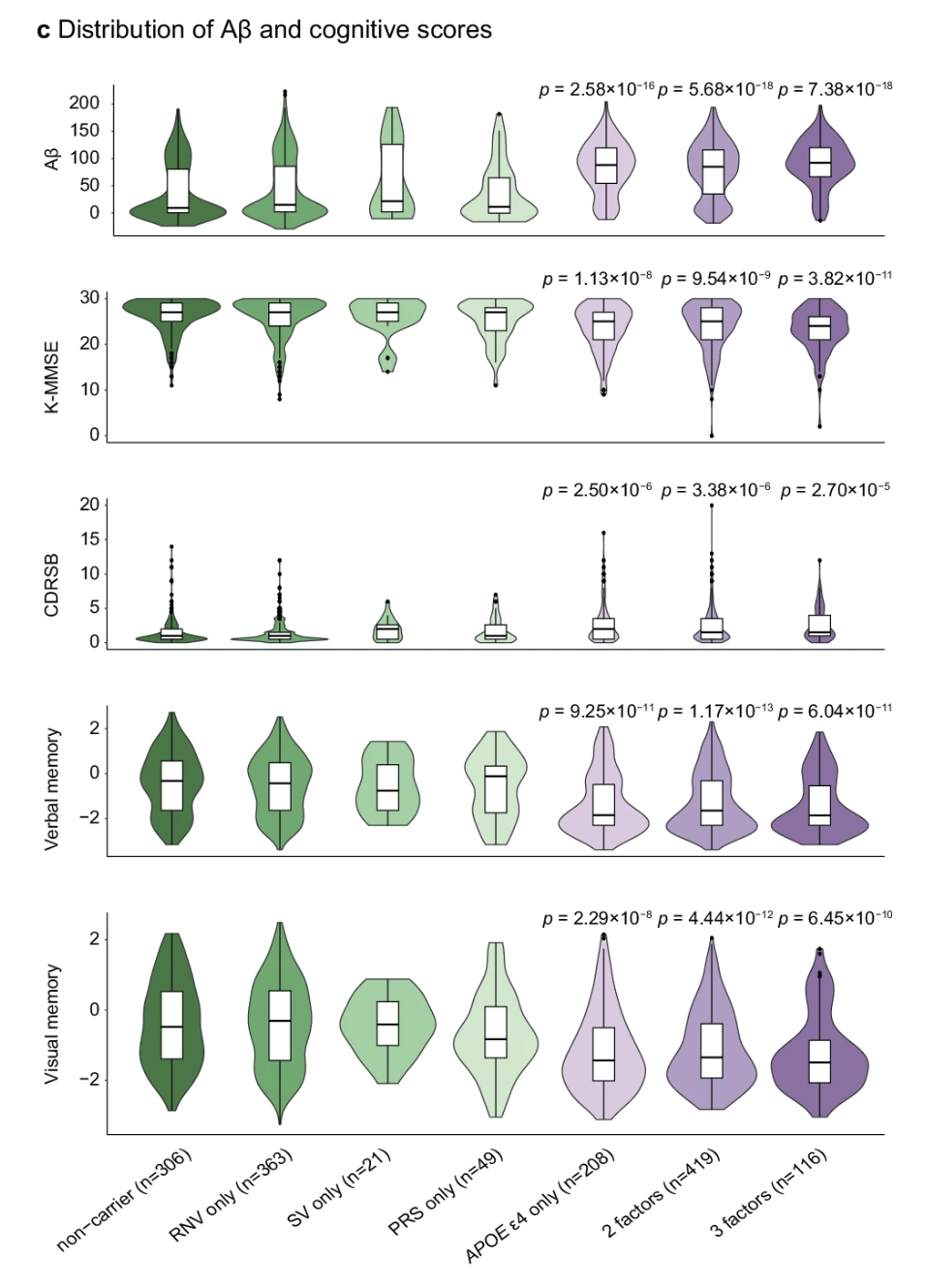
Figure: Genetic risk accumulates in Alzheimer’s disease. This violin plot shows how the number of genetic risk factors affects Alzheimer’s disease outcomes. Individuals are grouped by their risk burden: non-carriers (no risk factors), single-factor carriers (rare noncoding variants only, structural variants only, high polygenic risk score only, or APOE ε4 only), and those with 2 or 3 risk factors combined. As the number of risk factors increases from left to right, several patterns emerge: amyloid-β (Aβ) burden increases, cognitive function declines (lower K-MMSE scores), dementia severity worsens (higher CDR-SB scores), and both verbal and visual memory deteriorate. All comparisons show statistically significant trends (p-values displayed above each panel). This demonstrates that genetic risk is cumulative—having multiple risk factors (whether common variants, rare variants, structural variants, or APOE ε4) compounds disease severity more than any single factor alone. This is composite genetic architecture in action. Source: K-ROAD consortium (2025). Integrated analysis of common and rare genetic variants identifies multiple loci associated with Alzheimer’s disease. Nature Communications. https://www.nature.com/articles/s41467-025-59949-y. License: CC-BY.
Example 3: Autism Spectrum Disorder (ASD)
Autism spectrum disorder (ASD) is perhaps the most genetically heterogeneous condition we’ll discuss. It’s not one disorder—it’s a spectrum of neurodevelopmental differences with diverse genetic causes.
De Novo Variants
Many cases of autism—especially those classified as level 3 (requiring very substantial support)—are associated with de novo variants: new mutations that weren’t inherited from either parent. These include:
- De novo protein-truncating variants (dnPTV): mutations that create premature stop codons
- De novo copy-number variants (dnCNV): large deletions or duplications
These variants often hit genes involved in brain development, especially during fetal and early postnatal periods. Each individual mutation is rare, but collectively they account for a meaningful fraction of autism cases.
Inherited Rare Variants
Many people with autism carry inherited rare variants—missense mutations or regulatory variants passed from parents who may not have autism themselves. Each variant might slightly increase risk without being sufficient to cause the disorder on its own.
This is sometimes called the “additive rare-variant model.” You inherit a few risk variants from each parent, they add up, and if you cross a threshold, you develop ASD.
Polygenic Risk
Common SNPs explain about 30–40% of population-level liability for autism (Al-Beltagi et al. 2024, World J Clin Pediatr). These are the typical GWAS variants—thousands of common alleles with tiny effects. As with the other disorders, polygenic risk modulates the impact of rare variants.
Syndromic Autism
Some autism cases occur as part of known genetic syndromes:
- Fragile X syndrome (mutation in FMR1)
- Rett syndrome (mutation in MECP2)
- Tuberous sclerosis (mutations in TSC1 or TSC2)
In these cases, autism features are just one part of a broader clinical picture. These syndromic cases are easier to diagnose genetically, but they’re a minority of all ASD.
Sex Differences and the Female Protective Effect
Autism is diagnosed about 4 times more often in males than in females. One explanation is the female protective effect: females appear to require a higher genetic burden to develop autism. In other words, females can carry more risk variants without crossing the threshold into clinical diagnosis. When females do develop autism, they often carry more rare variants or a higher polygenic score than affected males.
This sex difference might be biological (X-linked genes, hormonal effects) or diagnostic (females with autism might be underdiagnosed because they present differently).
Functional Convergence
Despite involving hundreds of different genes, autism-associated variants converge on specific biological processes:
- Neuronal development (how neurons form and migrate during brain development)
- Synaptic signaling (how neurons communicate at synapses)
- Chromatin regulation (how DNA is packaged and which genes are turned on or off)
Many of these genes are especially active during mid-fetal cortical development—the period when the cortex is forming. This suggests that autism often arises from disruptions during a specific developmental window (Yuen et al. 2024, Cell Genom).
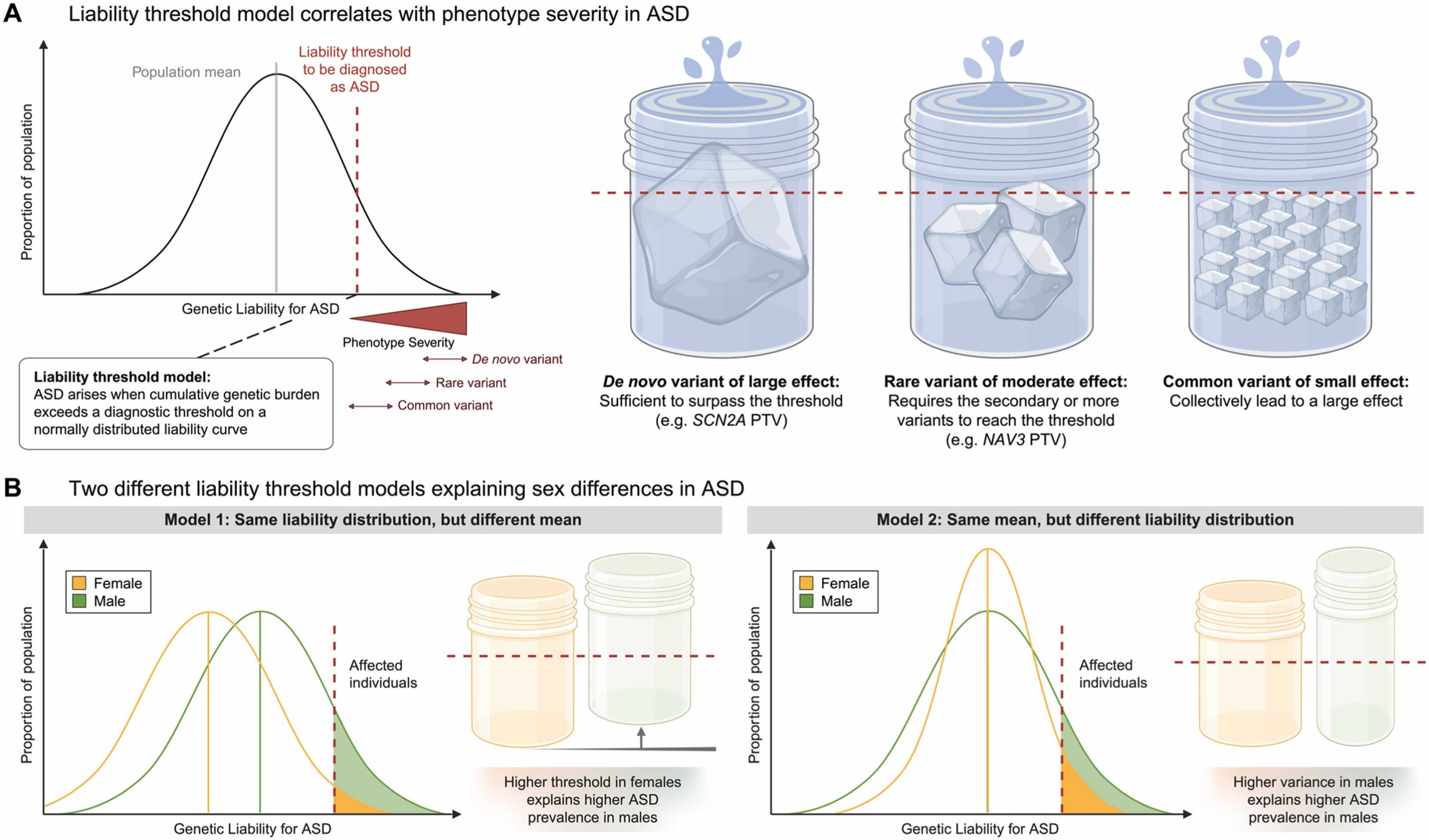
Figure: Multiple genetic contributors to autism risk. This figure shows how different types of genetic variants each contribute to ASD liability. De novo variants (both CNVs and PTVs), inherited rare variants, polygenic risk from common SNPs, and X-linked variants all add to the total genetic burden. No single category explains all cases—autism has a truly composite architecture where multiple variant types combine to determine individual risk. Source: Kim, Y.S. et al. (2025). Autism spectrum disorder: An evolutionary perspective. Molecules and Cells. https://doi.org/10.1016/j.mocell.2025.100248 License: CC-BY.
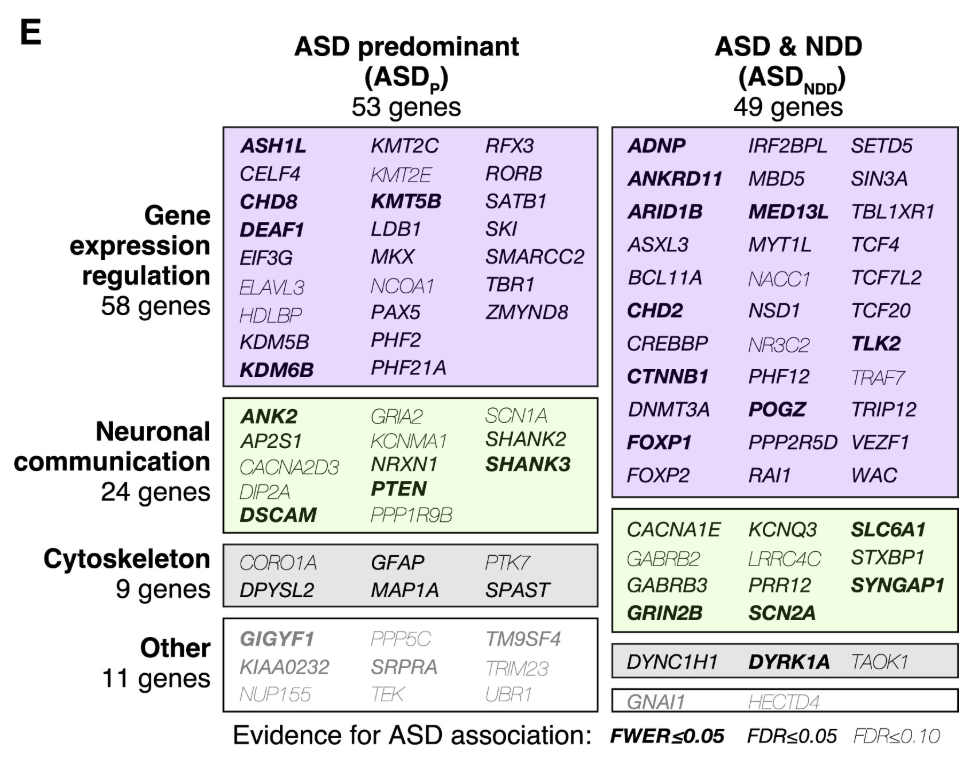
Figure: Network of ASD-associated genes converging on synaptic and chromatin pathways. Despite genetic heterogeneity, most ASD-associated genes interact in common functional networks. The major hubs are synapse formation (how neurons connect), chromatin regulation (how genes are turned on and off), and synaptic transmission (how neurons communicate). Different genes disrupt different nodes in these networks, but they all affect the same core processes. Many roads, but they lead to the same destination. Source: Satterstrom, F.K. et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. https://www.cell.com/cell/fulltext/S0092-8674(19)31398-4. License: CC-BY.
Genetic Heterogeneity and Biological Convergence
Let’s step back and see the pattern. All three disorders—DCM, Alzheimer’s, and autism—show genetic heterogeneity: different patients have different causal genes or variants leading to clinically similar outcomes.
In DCM, one patient might have a TTN mutation, another might have high polygenic risk, and a third might have multiple rare variants in different genes. Yet they all end up with a dilated, weakened heart.
In Alzheimer’s, one patient might carry APOE ε4/ε4, another might have a rare TREM2 variant, and a third might have high polygenic risk. But they all accumulate amyloid plaques and lose neurons.
In autism, one child might have a de novo CNV, another might inherit multiple rare variants, and a third might have high polygenic risk. But they all show differences in social communication and repetitive behaviors.
This is the paradox of complex diseases: many genetic routes, but shared clinical outcomes.
How is this possible? Because of biological convergence. Despite the genetic diversity, the variants funnel into common pathways:
- In DCM: sarcomere structure, cell adhesion, ECM
- In Alzheimer’s: microglial clearance, lipid metabolism, amyloid processing
- In Autism: synaptic signaling, chromatin regulation, neuronal development
Think of it like a river system. Many small streams start in different places—some in mountains, some in forests, some in plains. But they all flow into the same major rivers, which eventually reach the ocean. The starting points are heterogeneous, but the end point converges.
Features of Complex Genetic Architecture
Let’s summarize the key features that make genetic architecture complex:
Locus heterogeneity: Different genes can cause the same phenotype. In DCM, mutations in TTN, LMNA, DSP, or FLNC all lead to heart failure. The genes are different, but the outcome is similar.
Allelic heterogeneity: Different mutations in the same gene can have different effects. A missense mutation in TTN might cause mild DCM, while a truncating mutation causes severe early-onset disease. Even within one gene, there’s variability.
Temporal context: When does the gene act? Some genes matter during fetal development (like many autism genes). Others act in adulthood (like Alzheimer’s risk genes). The timing shapes the phenotype.
Sex differences: Males and females can have different genetic susceptibilities. The female protective effect in autism is one example. Sex hormones, X-chromosome dosage, and other factors modulate genetic risk.
Penetrance and modifiers: Having a pathogenic mutation doesn’t guarantee disease. Penetrance—the probability that a genotype leads to the phenotype—is influenced by polygenic background, other rare variants, and environmental factors.
Environmental interactions: Genetics doesn’t act in a vacuum. Diet, infections, toxins, stress, and lifestyle all interact with genetic risk. In Alzheimer’s, vascular health modifies genetic risk. In DCM, viral infections or alcohol use can trigger disease in genetically susceptible people.
Pathway convergence: This is the flip side of genetic heterogeneity. Diverse variants converge on shared biological pathways. It’s what allows clinically similar outcomes despite genetic diversity.
So genetic architecture isn’t a simple blueprint. It’s a dynamic landscape where multiple factors interact. Different people reach the same disease endpoint through different paths, and the same genetic variant can have different effects depending on the context.
Why This Matters
Understanding genetic architecture is not just an academic exercise. It has real implications for how we interpret genetic tests, counsel patients, and develop treatments.
Interpreting genetic tests: If a patient has DCM and you find a TTN mutation, you can’t just say “case closed.” You also need to consider their polygenic risk. A TTN carrier with low PGS might never develop symptoms, while one with high PGS might develop severe disease early. The mutation is one piece of the puzzle, not the whole answer.
Genetic counseling: When you tell someone they carry a risk variant, context matters. Are they in a high polygenic risk group? Do they have other rare variants? What’s their family history and environment? All of this shapes their actual risk.
Drug development: If multiple genetic variants converge on the same pathway—like microglial efferocytosis in Alzheimer’s—that pathway becomes an attractive drug target. You don’t need to fix every individual variant. You just need to boost the impaired pathway.
Precision medicine: Understanding genetic architecture allows personalized risk prediction and treatment. If someone has high polygenic risk for DCM, you can monitor them closely and intervene early. If someone has a rare variant plus high polygenic risk, they might need more aggressive treatment.
Putting It All Together
Let’s recap what we’ve learned about genetic architecture.
Most traits and disorders have composite genetic architectures that combine rare variants, common variants, and modifying factors. It’s not one gene, one disease. It’s many genes, many variants, interacting in complex ways.
The same clinical diagnosis can result from different genetic causes. This is genetic heterogeneity. In DCM, some patients have rare pathogenic mutations, others have high polygenic risk, and still others have multiple rare variants. Yet they all present with similar symptoms.
Despite this heterogeneity, many genetic variants converge on shared biological pathways. In DCM, it’s muscle structure and ECM. In Alzheimer’s, it’s microglial clearance and lipid metabolism. In autism, it’s synaptic signaling and chromatin regulation. This functional convergence explains how diverse genetic routes produce similar clinical outcomes.
Effect size, allele frequency, sex, developmental timing, and environment all influence disease expression. A variant’s impact depends on context. When does it act? In which tissues? In males or females? Against what polygenic background? With what environmental exposures? All of these factors modulate genetic risk.
Grasping this complexity is essential for interpreting genomic results and advancing personalized medicine. We can no longer think in simple Mendelian terms for most diseases. We need to embrace the complexity—the heterogeneity, the interactions, the context-dependence—to truly understand how genetics shapes health and disease.
Genetic architecture is messy. But it’s also beautiful, because it shows us how the same biological systems can be disrupted in many different ways, yet still funnel into coherent disease mechanisms. That gives us hope: if we understand the convergent pathways, we can develop therapies that work across genetically diverse patients. We don’t need to fix every mutation—we just need to fix the shared pathway.