human genetics
Chapter 14. Additive and Dominant Alleles
So far, we’ve talked about how mutations can disrupt genes and cause disease. But here’s something interesting: not all genetic variants work the same way. Some variants are like a light switch—flip it once, and the whole room changes. Others are more like dimmer switches—you need to adjust many of them to see a real difference.
Let’s use height as our example. Height is something we can all relate to, and it turns out genetics influences it in two very different ways. We’ll look at dominant alleles—the “light switch” kind—that cause dramatic changes with just one mutated copy. Then we’ll explore additive alleles—the “dimmer switch” kind—where hundreds or even thousands of genetic variants each nudge your height up or down by tiny amounts.
To see dominant alleles in action, we’ll examine two genetic disorders: Achondroplasia (which causes much shorter stature) and Marfan syndrome (which causes much taller stature). Then we’ll contrast these with how height works in most people, where common and rare variants team up to create the familiar bell curve of human heights. All heights here are in centimeters (cm), the standard unit for medical measurements.
Dominant Alleles and Achondroplasia
Achondroplasia is the most common form of dwarfism, affecting about 1 in every 15,000 to 25,000 babies born. The name literally means “without cartilage formation,” which is a bit misleading. People with achondroplasia can form cartilage just fine—the problem is what happens next.
How It Works
Think of your bones as having two phases of development. First, your body lays down a cartilage template (cartilage is that tough but flexible tissue you can feel in your nose and ears). Then, during childhood and adolescence, that cartilage gradually converts into hard bone through a process called ossification. In achondroplasia, this conversion process gets disrupted, especially in the long bones of your arms and legs.
The result? Without treatment, adult males with achondroplasia average about 131 cm tall (roughly 4 feet 4 inches), while adult females average 124 cm (about 4 feet 1 inch). Compare that to average heights of around 175 cm for men and 162 cm for women in many populations.
But height isn’t the only feature. People with achondroplasia typically have:
- An average-sized torso (their trunk is normal length)
- Short arms and legs, with the upper arms and thighs especially shortened—doctors call this “rhizomelia”
- Limited ability to fully straighten their elbows
- A larger-than-average head (macrocephaly) with a prominent forehead
- Short fingers, where the ring and middle fingers sometimes spread apart, giving the hand a “trident” (three-pronged) appearance
Here’s something that might surprise you: about 80% of achondroplasia cases pop up out of nowhere. The parents have completely normal stature, and the mutation happens brand new (we call these de novo or sporadic mutations) in the egg or sperm that made that child (Ornitz et al. 2017, Dev Dyn).
The Genetic Culprit: FGFR3
Achondroplasia is caused by mutations in a gene called FGFR3, which sits on the short arm of chromosome 4. FGFR3 stands for “Fibroblast Growth Factor Receptor 3”—quite a mouthful, but the name tells you what it does. This gene makes a receptor protein that sits on the surface of cartilage cells (chondrocytes) in your growth plates. Growth plates are the zones of growing tissue near the ends of your long bones—they’re what make you taller during childhood.
Now here’s the kicker: nearly everyone with achondroplasia has the exact same mutation. It’s a single amino acid swap—glycine gets replaced by arginine at position 380 of the protein. Scientists write this as p.Gly380Arg (or G380R for short). This one-letter change in the genetic code shows up in achondroplasia patients across different populations worldwide. It’s like finding the same typo in books printed on different continents—it tells us this particular mutation is the main troublemaker.
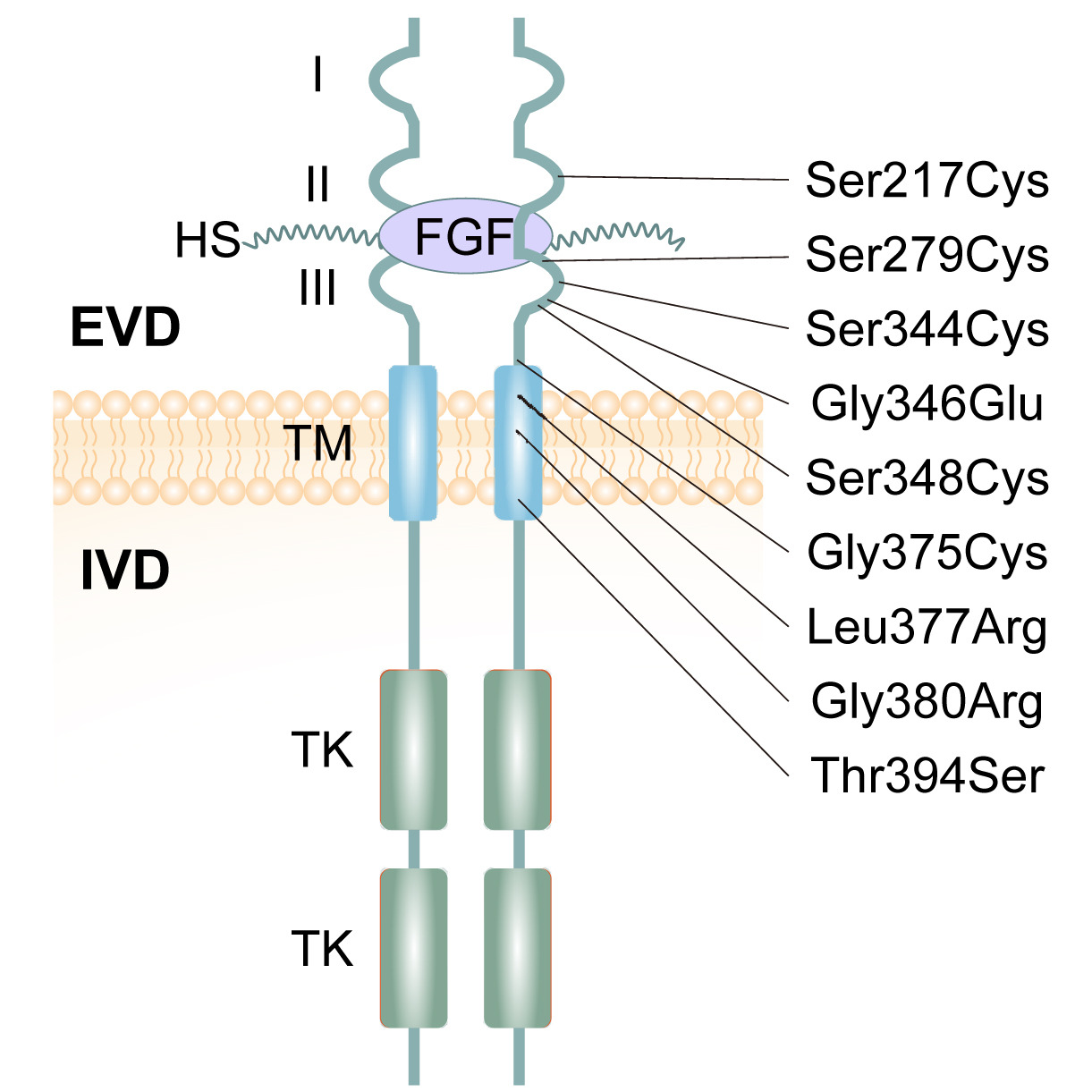
Figure: FGFR3 receptor structure and achondroplasia mutation sites. The FGFR3 receptor spans the cell membrane. Outside the cell, the extracellular domain (ECD) contains three immunoglobulin-like regions (I, II, III) that can bind to growth factors and heparan sulfate (HS). The transmembrane domain (TM) anchors the receptor in the membrane—this is where the G380R mutation is located. Inside the cell, the intracellular domain (ICD) contains tyrosine kinase (TK) regions that send signals when the receptor is activated. Most achondroplasia mutations cluster in the TM region, right where G380R sits. Source: Sobreira, N. et al. (2024). Achondroplasia. HealthMedicine. https://www.sciencedirect.com/science/article/pii/S2352304224002332. License: Creative Commons.
Why This Mutation Causes Problems
Normally, FGFR3 acts like a brake on bone growth. When growth factors bind to the receptor, two receptor molecules come together (dimerize), activate each other, and send a signal into the cell saying “slow down bone growth.” This is actually healthy—it prevents your bones from growing too much.
But the G380R mutation changes everything. This mutation sits in the transmembrane domain—the part of the receptor buried in the cell membrane. The swap from glycine (a small, flexible amino acid) to arginine (a much larger one with a bulky side chain) makes the receptor behave differently. It promotes dimerization even when no growth factor is around (Hartl et al. 2022, bioRxiv).
Think of it like a car alarm that goes off even when nobody’s trying to break in. The receptor is stuck in the “on” position, constantly telling growth plate chondrocytes to slow down—even when they should be growing normally. The receptor activates signaling pathways (MAPK and STAT1) that suppress chondrocyte proliferation and maturation. Fewer dividing chondrocytes and less cartilage production means less bone growth, especially in the long bones.
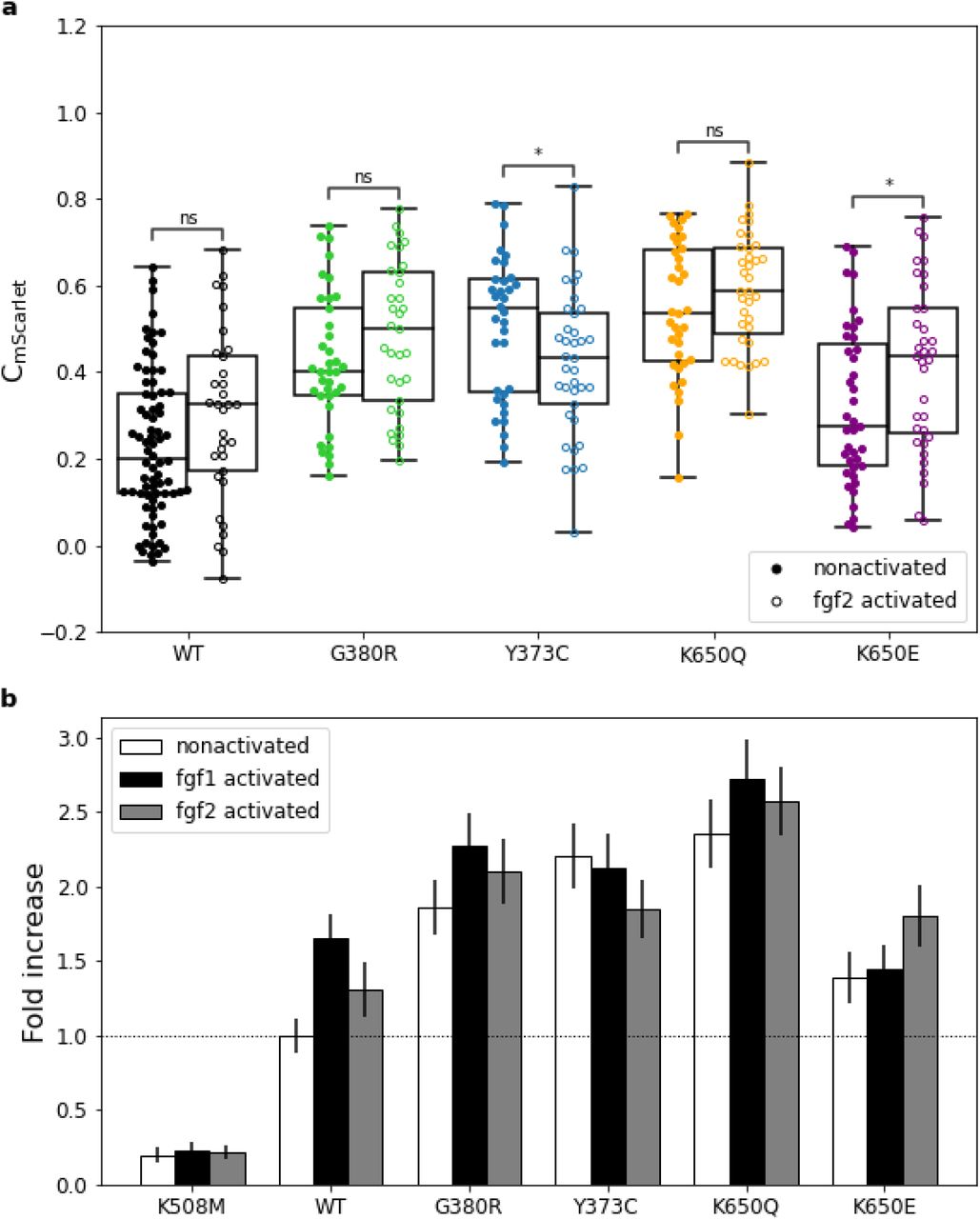
Figure: G380R mutation increases FGFR3 signaling activity. This graph shows how strongly different FGFR3 variants activate downstream signaling, measured by GRB2 (an adaptor protein) recruitment. Higher bars indicate stronger signaling. The G380R variant shows elevated activation even without adding FGF ligand—the receptor signals constitutively, which doesn’t occur with the wild-type receptor. This ligand-independent activation explains why one mutated copy is sufficient to cause achondroplasia. Source: Hartl, D. et al. (2022). Molecular mechanisms underlying pathogenic FGFR3 mutations. bioRxiv. https://www.biorxiv.org/content/10.1101/2022.04.11.487861v1.full License: CC-BY-NC-ND 4.0.
Why It’s Dominant
Here’s the key: achondroplasia is a dominant disorder. You only need one mutated copy of FGFR3 to have the condition. Even though you also have one perfectly normal copy inherited from your other parent, that normal copy can’t compensate. The mutant receptor is so overactive that it dominates the situation—hence “dominant allele.”
Dominant Alleles and Marfan Syndrome
Let’s look at the flip side: a dominant disorder that makes people taller instead of shorter. Marfan syndrome is an autosomal dominant disorder affecting connective tissue—the material that holds your body together. People with Marfan syndrome are usually much taller than average: adult males often exceed 190 cm (about 6 feet 3 inches), while females typically reach 175–180 cm (5 feet 9 inches to 5 feet 11 inches) or more.
But tall stature is just one piece of the puzzle. Marfan syndrome comes with a distinctive set of features:
- Long, slender limbs and fingers (doctors call the long fingers “arachnodactyly”—literally “spider fingers”)
- Lens dislocation in the eye (ectopia lentis)—the lens can slip out of place
- A predisposition to thoracic aortic aneurysms—dangerous bulges in the body’s main artery that can rupture
Unlike achondroplasia, which shortens the limbs while keeping the trunk normal, Marfan syndrome elongates body proportions overall (Sakai et al. 2016, Genes).
The Genetic Culprit: FBN1
Marfan syndrome is caused by mutations in the FBN1 gene on chromosome 15. FBN1 encodes a protein called fibrillin-1, which is a structural protein. Think of fibrillin-1 as microscopic ropes that form the scaffolding of connective tissue. These “ropes” (microfibrils) provide strength and elasticity to tissues like the aorta, the ligaments that hold your eye lens in place, and the connective tissue in your bones.
Unlike achondroplasia—where nearly everyone has the same mutation—Marfan syndrome is caused by over 1,000 different mutations in FBN1. These include missense mutations (amino acid swaps), nonsense mutations (premature stop signals), splice-site mutations (errors in how the gene is processed), and small insertions or deletions. Many of these mutations hit either the calcium-binding EGF-like domains or the 8-cysteine domains of fibrillin-1, disrupting the protein’s structure or how it folds (Learn.Genetics Utah).
See Figure: Fibrillin-1 structure from Learn.Genetics Utah. Fibrillin-1 proteins assemble into long, strong fibers that reinforce connective tissue throughout the body. When there is insufficient fibrillin-1, or when the protein is misshapen, the scaffolding is weak—this is what happens in Marfan syndrome.
Why FBN1 Mutations Cause Tall Stature
FBN1 mutations are dominant alleles—one bad copy is enough to cause Marfan syndrome. But how do defective microfibrils lead to excessive height?
It turns out fibrillin-1 has a second job beyond structural support: it regulates a signaling molecule called TGF-β (transforming growth factor beta). Normally, fibrillin-1 microfibrils capture and sequester TGF-β, keeping it in check. When fibrillin-1 is defective, TGF-β escapes this regulation and becomes overactive.
Excessive TGF-β signaling promotes abnormal growth in connective tissues, including bones. It drives long bone elongation beyond normal limits, resulting in the tall stature and elongated limbs characteristic of Marfan syndrome. So even though you have one normal FBN1 copy, the defective copy disrupts the scaffolding and unleashes too much TGF-β—enough to override the normal copy’s function. That’s why it’s dominant.
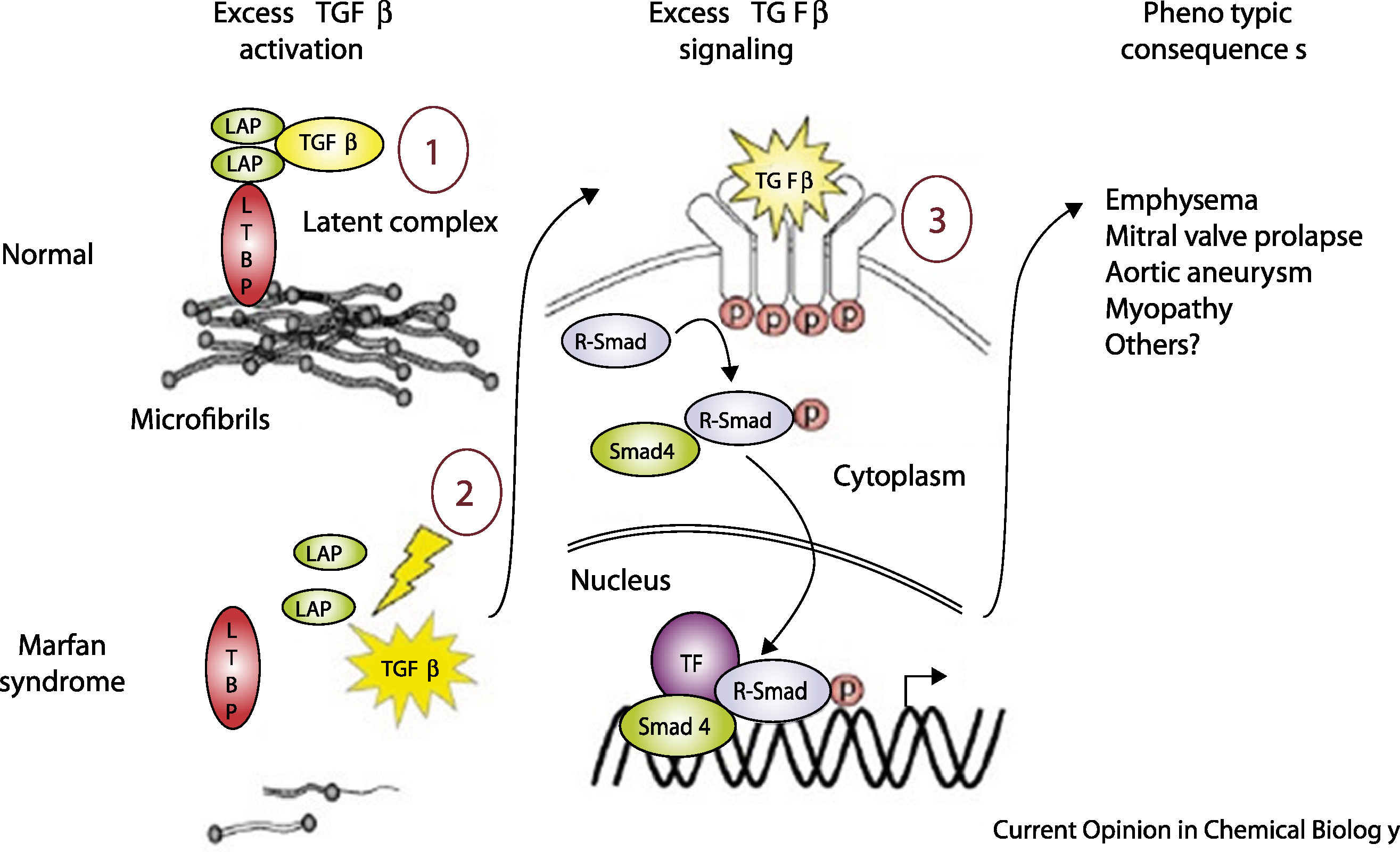
Figure: TGF-β dysregulation in Marfan syndrome. Left: In healthy tissue, fibrillin-1 microfibrils sequester TGF-β by binding to latency-associated peptide (LAP) and latent TGF-β binding protein (LTBP), keeping TGF-β signaling in check. Right: When microfibrils are deficient (as in Marfan syndrome), TGF-β is released and activates its receptors. This triggers excessive signaling through Smad proteins and transcription factors, driving abnormal tissue growth and the skeletal features of Marfan syndrome. Source: Coelho, S.G. & Almeida, A.G. (2020). Marfan syndrome revisited: From genetics to the clinic. Cellular Signalling. https://www.sciencedirect.com/science/article/pii/S217420492030115X. License: Creative Commons.
Additive Alleles and Normal Variation in Human Height
Now let’s shift gears. We’ve just seen how single dominant mutations in FGFR3 or FBN1 can dramatically alter height. But here’s the thing: most people don’t have achondroplasia or Marfan syndrome. For the vast majority of us, height is influenced by additive alleles—lots of genetic variants, both common and rare, each contributing a tiny nudge up or down.
Instead of one mutation flipping a switch, imagine thousands of little dials all adjusting your height slightly. Some dial you a bit taller, others a bit shorter. The cumulative effect of all these dials determines where you land on the height spectrum. This is why human height follows a normal distribution—a bell curve—with most people clustering around the average and fewer people at the very short or very tall extremes.
The Genetic Basis of Height Variation
Let’s look at some numbers to appreciate just how polygenic (literally “many genes”) height really is.
A massive genome-wide association study (GWAS) analyzed 5.4 million people from diverse ancestries (Yengo et al. 2022, Nature). The study identified 12,111 independent single-nucleotide polymorphisms (SNPs)—single-letter DNA variants—associated with height. Almost all of these SNPs are common variants, meaning they appear in more than 1% of the population (we abbreviate this as MAF > 1%, where MAF stands for “minor allele frequency”).
These 12,111 SNPs are scattered across 7,209 different genomic regions, covering about 21% of the entire genome. Together, they explain most of the height variation we can detect using common SNPs. Interestingly, genes like FGFR3 and FBN1 show up in this list—but as common variants with subtle effects, not the severe mutations causing achondroplasia or Marfan syndrome. Other genes in the list regulate growth-plate function and connective-tissue development, just like the Mendelian examples.
But common variants aren’t the whole story. A complementary study used whole-genome sequencing on 25,465 unrelated Europeans to explore the role of rare variants—those with frequencies below 0.1% (Wainschtein et al. 2022, Nat Genet). SNP-based heritability of height was estimated at 0.68 (with a standard error of 0.10), meaning about 68% of height variation can be attributed to genetic differences.
Here’s where it gets interesting. Rare variants contribute substantially, but how they contribute depends on something called linkage disequilibrium (LD). LD measures how tightly linked a variant is to nearby variants. Low-LD rare variants—those that aren’t correlated with nearby common variants—accounted for 0.31 (31%) of the phenotypic variance. In contrast, high-LD rare variants—those tightly linked to common variants already captured by GWAS—contributed only 0.03 (3%).
What does this mean? Low-LD rare variants provide new, independent genetic information that GWAS can’t easily pick up. High-LD variants, on the other hand, are essentially “tagging along” with common variants we already knew about, so they don’t add much new information. These rare variants are often protein-altering changes under negative selection (evolution weeds them out because they’re slightly harmful), but they still contribute additively, not dominantly.
How Additive Alleles Work
Let’s make this concrete with an example. Imagine you inherit a height-increasing SNP that adds 0.2 cm to your height. You also inherit a height-decreasing SNP that subtracts 0.15 cm. Another SNP adds 0.1 cm. Another subtracts 0.05 cm. And so on, thousands of times.
Your final height is the net sum of all these tiny increments and decrements, plus environmental factors like nutrition and health during childhood. Because each individual SNP has such a small effect, you need to add up thousands of them to explain someone’s height.
This additive model creates the familiar bell curve. Most people inherit a mix of height-increasing and height-decreasing variants that roughly balance out, putting them near the population average. A few people inherit more height-increasing variants than average and end up taller. A few inherit more height-decreasing variants and end up shorter. But nobody’s height is determined by a single dominant mutation—unless they have a Mendelian disorder like achondroplasia or Marfan syndrome.
Rare variants enhance this picture by capturing additional variance that common SNPs miss. But crucially, they still work additively. Even if you have a rare protein-altering variant in, say, a growth-related gene, it doesn’t override your other variants the way a dominant mutation would. It just adds its small effect to the pile.
Comparing Dominant and Additive Alleles
Let’s put everything together. We’ve looked at three scenarios:
Achondroplasia (Dominant): Caused by mutations in the FGFR3 gene on chromosome 4, almost always the p.Gly380Arg substitution. This single mutation is enough to cause short stature—about 131 cm in adult males and 124 cm in females. The mutation makes the FGFR3 receptor overactive, suppressing growth plate chondrocyte proliferation. It’s a dominant allele: one copy causes the full effect, and penetrance is complete (everyone with the mutation has the disorder).
Marfan syndrome (Dominant): Caused by over 1,000 different mutations in the FBN1 gene on chromosome 15. These mutations disrupt fibrillin-1 microfibrils, weakening connective tissue and unleashing excessive TGF-β signaling. The result is tall stature—often above 190 cm in males—plus other features like lens dislocation and aortic aneurysms. Like achondroplasia, it’s a dominant allele: one bad copy is enough, and penetrance is complete.
Normal height variation (Additive): Influenced by thousands of variants across multiple genes—including FGFR3, FBN1, and many others. The 2022 GWAS identified 12,111 common SNPs (MAF > 1%) spread across 7,209 genomic regions. Rare variants (MAF < 0.1%), especially those in low LD, contribute an additional 31% of the phenotypic variance. Each variant nudges height up or down by a tiny amount—fractions of a centimeter. Your height is the cumulative sum of all these effects. Most people end up somewhere between 157 cm and 183 cm (roughly 5 feet 2 inches to 6 feet), depending on ancestry and sex. These are additive alleles: small effects that add up, with variable penetrance depending on how many you inherit.
The contrast is striking. Dominant alleles like those in achondroplasia and Marfan syndrome act like master switches, overpowering the normal copy and producing a predictable phenotype. Additive alleles, on the other hand, are like a committee—each member gets a tiny vote, and the outcome depends on the majority decision across thousands of votes.